前言
合成生物学在医药、化妆品、农业、食品等各个行业都有广阔的应用场景,而每一具体行业项下的合规监管都各具特色。除本系列第二篇《合成生物学合规及投融资法律指南(二)——一般性合规》所提及的一般性合规监管外,合成生物学企业——特别是产品型公司——还需进一步关注其产品/服务所在的具体行业的现行监管规范。本文将从合成生物学在医药行业的应用角度出发,介绍合成生物学技术在这个行业的发展现状及合规监管情况。
合成生物学产业化的发展历程中,其在医药行业的应用是合成生物学产业化的肇始[1],也是研发资金投入最多的方向之一。其本质原因是人类希望通过合成生物学解决人类最终极的需求——健康和长寿。合成生物学目前在医药领域的应用包括细胞治疗、基因治疗、活体生物药、原料药、医药中间体等诸多方向。
一、合成生物学在医药行业的发展概览
如本系列第一篇《合成生物学合规及投融资法律指南(一)——概览》所描述,随着基因编辑、“Design-Build-Test-Learn”(DBTL)流程和工程生物学等技术和工艺的发展,合成生物学在欧美国家的生物医药产业中进入了高速成长期。根据相关数据显示[2],2010年至今,全球发表合成生物学领域与药物研发有关的文献呈逐年递增趋势,利用合成生物技术开发药物正处于迅速发展阶段。其中,美、中、法的主要研究及应用方向为生物医药、生物能源和环境修复,英、德、瑞士为生物医药、环境修复和化工制品合成。作为合成生物学研究的热点和重点,结合生物医药领域的研究在各国的合成生物学研究领域中均占首位,主要涉及天然产物药物的挖掘与生物合成、细胞及基因治疗、遗传线路设计与疾病诊断等研究[3]。
从对人体的不同作用机制来看,广义而言,合成生物学技术在医药行业的主要应用包括:
-
细胞治疗。即使用人体细胞进行治疗(无论是否经基因修饰),具体分为免疫细胞治疗、干细胞治疗及其他体细胞治疗。细胞疗法中最有名的应用,当属以CAR-T疗法为代表的免疫细胞治疗。严格来讲,CAR-T疗法也属于体外基因治疗的一种,其原理是对人体T细胞进行基因修饰,使其靶向消灭特定肿瘤细胞。
-
基因治疗。又名“基因编辑治疗”,通过编辑人体细胞中致病基因(包括修改、剪切或增补),从而达到治疗效果。根据基因编辑的实现方式,分为体内基因治疗(in vivo)和体外基因治疗(ex vivo)两种:
-
体内基因治疗:将外源基因或基因编辑工具(如CRISPR组件)通过载体导入人体,并直接编辑或增补人体细胞的基因,从而发挥治疗作用。例如,美国Broad Institute[4]旗下Editas Medicine研发的体内基因疗法EDIT-101中包括CRISPR/Cas9工具,预期可用于治疗Leber先天性黑蒙症10型(LCA10);
-
体外基因治疗:在人体外预先对细胞进行基因编辑,制备为经基因修饰的细胞或细胞衍生产品后再回输人体,从而发挥治疗作用。例如,美国Bluebird Bio研发的体外基因疗法Zynteglo,通过对患者体内分离的造血干细胞在体外进行基因修饰,使其回输患者体内生成正常的红细胞,从而治疗β地中海贫血症。
-
活体生物药。通过合成生物学技术改造微生物,从而研发基因修饰的活体生物药(live biotherapeutic products,LBPs),用于癌症、代谢疾病、肠道疾病等病症的治疗。
-
医药行业原材料(如原料药、医药中间体等)。此种方式更加偏向传统的合成生物学产业,不同的是其目标产品为生物制药行业的原材料。例如日前已向科创板递交招股书的弈柯莱生物,其主营业务之一即为向生物医药企业提供“西他列汀中间体”“度鲁特韦中间体”等生物医药中间体和前体,其与通化东宝(600867.SH)合作的中国首个使用生物合成技术生产的西格列汀仿制药获批,成为中国正式接受利用合成生物的方法制造、生产医药类产品的标志性事件[5]。
-
其他应用。包括小核酸药物、核苷类药物、mRNA药物/疫苗、circRNA药物/疫苗、重组胶原蛋白产品、抗HPV生物蛋白产品、氨基酸及其衍生物产品等。
二、合成生物学在医药行业落地的监管
根据上述分析,可知合成生物学在我国医药行业的应用涉及细胞及基因治疗、活体生物药(微生物治疗)、传统药品及原材料、医疗器械、化学品等多个具体领域。由于我国对于药品、医疗器械、化学品分别实行不同的监管制度,因此企业需针对性地了解各类产品所涉领域的监管法律。随着近年我国对于医药行业的监管趋向于完善和严格[6],保持对相关法律动态的追踪对于相关企业的重要性不言自明。
1. 细胞治疗及基因治疗(CGT)——以CAR-T为例
根据国家药监局(NMPA)发布的相关指导原则及相关实践,细胞及基因治疗(Cell and Gene Therapy, “CGT”)的分类如下图所示。虽然在业界认知中,从事细胞治疗及基因治疗的企业(“CGT企业”)属于生物创新药企业,而非传统意义上的合成生物学企业(后者以选品和量产为核心壁垒),但基于其底层技术相通、互相转型容易[7]等原因,我们仍对CGT企业的合规要点进行简要介绍。
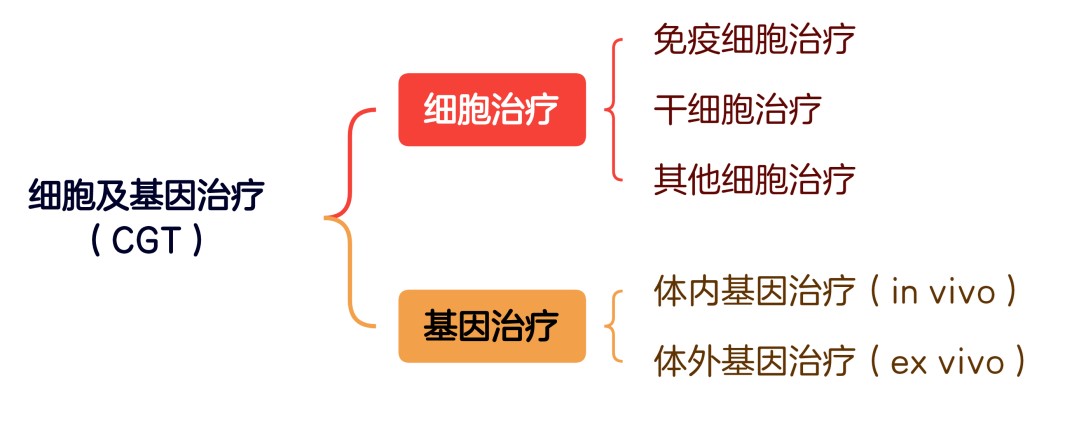
▲我国细胞及基因治疗(CGT)产品的分类
截至2021年,获得我国NMPA批准的CGT产品仅有四款[8],其中两款为CAR-T产品。CAR-T疗法/产品的原理是通过合成生物学技术,将能够辨识癌细胞抗原的嵌合抗原受体(chimeric antigen receptor,CAR)基因插入人体T细胞,并将其改造成CAR-T细胞,以实现识别并靶向消灭癌细胞的目的。由于CAR-T疗法/产品同时属于细胞治疗和基因治疗范畴,因此我们将以此为例来介绍相关CGT企业的主要合规要点。
-
细胞治疗监管的“双轨制”。我国目前对细胞治疗仍采取“双轨制”监管。具体而言,作为“医疗技术”的细胞治疗由国家卫健委监管,经医疗卫生机构批准立项后,方可作为临床研究在机构内部实施,且在“魏则西事件”后其临床应用进一步收紧;作为“药品”的细胞治疗产品,则需根据NMPA的监管规则开展临床研究、注册申请和产业化生产。虽然普遍认为“双轨制”还将继续存在一段时间,但鉴于CGT企业无法参与医疗卫生机构内部的细胞治疗临床研究,因此细胞治疗产品商业化的唯一途径是作为药品申报并接受监管。2022年1月,NMPA发布的新版《药品生产质量管理规范—细胞治疗产品附录(征求意见稿)》也提及了比照药品监管的细胞治疗产品与普通药品的不同,并提出了相关企业应遵守的各种具体要求。
-
CAR-T产品的属性。近年来,NMPA针对CGT的临床试验及非临床研究出台了一系列指导原则,主要包括《细胞治疗产品研究与评价技术指导原则(试行)》(2017年12月)、《免疫细胞治疗产品临床试验技术指导原则(试行)》(2021年2月)、《基因治疗产品药学研究与评价技术指导原则(征求意见稿)》(2020年9月)、《基因修饰细胞治疗产品非临床研究技术指导原则(试行)》(2021年11月)、《基因治疗产品非临床研究与评价技术指导原则(试行)》(2021年11月)、《体内基因治疗产品药学研究与评价技术指导原则(试行)》(2022年5月)等。根据上述指导原则,NMPA按照分类管理原则对CGT产品进行了划分,而CAR-T产品同属“免疫细胞治疗产品”和“基因治疗产品(基因修饰细胞治疗产品)”,因此需要同时遵守两个细分领域的监管原则。
-
外资准入。根据《外商投资准入特别管理措施(负面清单)(2021年版)》,“人体干细胞、基因诊断与治疗技术开发和应用”为外商投资禁止类项目,但目前对上述表述的具体含义尚无明确的官方指引。在实践中,出于谨慎合规目的,有境外融资/上市需求的绝大部分CGT企业均搭建VIE协议控制结构,如永泰生物、药明巨诺、科济药业、亘喜生物等。虽然存在CAR-T疗法企业传奇生物(WFOE结构)和复星凯特(中外合资)的例外,但一般认为其为情况特殊的孤例,不具有普遍参考性。
-
人类遗传资源管理合规。CGT企业在其科研及临床研究中可能不可避免地涉及对我国人类遗传资源(“人遗资源”)的利用。根据《生物安全法》和《人类遗传资源管理条例》,外国组织、个人及其设立或者实际控制的机构(“外方单位”)不得在我国境内采集、保藏我国人遗资源,也不得向境外提供。如外方单位需要利用我国人遗资源开展科研活动的,应与我国科研/医疗机构、高校、企业合作,还应取得科技部下辖中国人类遗传资源管理办公室(“人遗办”)批准;但如果为获得药品/医疗器械在境内的上市许可在境内临床机构利用我国人遗资源开展国际合作临床试验、不涉及人遗资源出境的情形,则无需人遗办审批,备案即可。2022年3月,科技部发布的《人类遗传资源管理条例实施细则(征求意见稿)》对外方单位定义中的“实际控制”作了较为严格的解释,除传统的股权标准外,对决策/内部管理有重大影响、VIE控制也被明确纳入。如该细则正式出台,则①外方股东对决策/内部管理有重大影响(例如重大经营决策的一票否决权)的企业及②VIE协议控制结构中的内资公司均可能被认定为外方单位,其采集、保藏、利用我国人遗资源开展相关活动将受到较为严格的限制。
-
地方监管合规。CGT企业还应留意相关的地方监管规则。例如,上海药监局近期发布了《上海市自体嵌合抗原受体T细胞(CAR-T)治疗药品监督管理暂行规定》(2022年9月1日生效),该规定从机构人员、质量体系、生产工艺、设施设备、生物安全、留样管理、供应链管理、年度报告等方面对上海市的细胞治疗药品上市许可持有人、生产企业及其他相关单位制定了详细要求。
2. 活体生物药(LBPs)
美国食品药品监督管理局(“FDA”)将活体生物药(live biotherapeutic products,LBPs)定义为“含有活的微生物(如细菌)的药物,可以用于预防、处理、治疗人类疾病和适应症。其不包括疫苗、滤过性病毒、溶瘤细菌、基因治疗载体,一般也不会通过注射给药”[9]。
相较细胞及基因治疗(CGT),活体生物药的研究则更为前沿——目前尚无任何一款活体生物药产品获得美国FDA及中国NMPA的上市批准。当前在FDA和NMPA中推进速度最快的活体生物药分别为:(1) 辉凌制药(Ferring Pharmaceuticals)研发的用于治疗艰难梭菌感染(CDI)的活体生物药RBX2660,其有望在2022年内获得FDA批准其生物制剂许可申请(BLA);(2) 中国的知易生物研发的活体生物药SK08,其针对腹泻型肠易激综合征(IBS-D)的II期临床试验即将在2022年底完成。
活体生物药的制备并不必然需要利用合成生物学技术,比如上述提到的两款前沿活体生物药的相关菌株均非采用合成生物学技术获得。但是,合成生物学可以助力构建和筛选构建活体生物药所需的菌株,美国FDA也接受经基因重组的活体生物药申报。例如,美国Synlogic(NASDAQ:SYBX)开发的用于治疗苯丙酮尿症(PKU)的口服药SYNB1618(已获得美国FDA孤儿药及快速通道认定)及SYNB1934,其有效成分为经合成生物学改造的大肠杆菌Nissle 1917菌株,其作用机理为经基因修饰的菌株在患者体内表达产生苯丙氨酸氨裂合酶(PAL)并降低患者血液中的苯丙氨酸(Phe)水平。
针对利用合成生物学技术构建的活体生物药,除应遵守一般的药品监管以外,还应特别注意:(1) 相关从业者应严格遵守《生物安全法》,并根据主管部门后续出台的相关技术研究、开发活动风险分类标准及名录做好风险等级管理;(2) 遵守科技伦理原则,如未来合成生物学技术及其应用被认定为“科技伦理敏感领域”,则应根据相关要求,设立本单位的科技伦理(审查)委员会并严格履行伦理审查职责;(3) 根据NMPA《基因治疗产品非临床研究与评价技术指导原则(试行)》(2021年11月)的规定,利用合成生物学技术构建的活体生物药可能被归入“表达特定基因的基因修饰微生物”类型的基因治疗产品,从而需要遵守相关的研究规范及指导原则。
3. 药品监管(适用于原料药)
我国对药品的生产和销售的法律监管主要分为准入管理、质量规范管理、注册管理及流通管理四个层面,具体包括:
|
监管流程 |
具体要求 |
|
准入管理 |
|
|
质量规范管理 |
|
|
注册管理 |
|
|
流通管理 |
|
就新药申请而言,我们理解新生物药物的种类繁多,无论是天然生物药物还是基因工程药物、基因治疗药物,新药申报和审批程序大同小异,重点在于药物研究过程的不同。但如果新药已经通过审批,相关企业希望改用合成生物学的生产方式来提高效率或降低成本,在合规层面需重新进行GMP审批。
4. 医疗器械监管(适用于重组胶原蛋白产品、抗HPV生物蛋白产品)
我国目前对医疗器械实施分类管理,即基于对医疗器械的预期目的、结构特征、使用方法等因素的综合考量,将医疗器械按照风险程度分为三类管理,并从生产侧和经营侧分别进行监管[11]:
|
分类 |
风险因素 |
生产侧监管 |
经营侧监管 |
|
第一类 |
风险程度低,常规管理即可 |
向所在地设区的市级药品监管部门办理医疗器械生产备案 |
不需要许可和备案 |
|
第二类 |
具有中度风险,需要严格控制管理 |
应当经申请人所在地省、自治区、直辖市药品监管部门批准取得医疗器械生产许可证 |
由经营企业向所在地设区的市级人民政府药品监管部门办理备案并提交相关资料。但对产品安全性、有效性不受流通过程影响的第二类医疗器械,可以免于经营备案[12]。 |
|
第三类 |
具有较高风险,需要采取特别措施严格控制管理 |
应当向所在地设区的市级人民政府药品监管部门申请经营许可申请并提交相关资料 |
5. 安全生产监管(适用于医药中间体、氨基酸及其衍生物产品)
医药中间体,是医药化工原料至原料药或药品这一生产过程中的一种精细化工产品,化学药物的合成依赖于高质量的医药中间体。根据NMPA规定,医药中间体可视为药品原材料,无需按照药品规则生产报批、申请批号[13]。弈柯莱生物研发的西他列汀、度鲁特韦中间体以及川宁生物研发的青霉素类、头孢类中间体都属于医药中间体。基于此,我们认为医药中间体需遵守安全生产的相关监管规定,具体如下:
|
监管事项 |
具体要求 |
|
生产条件及许可 |
|
|
主要负责人、管理人员及机构职责 |
|
|
从业人员的教育、培训 |
|
|
生产安全事故责任、责任人及相关处理 |
|
医药中间体除需遵守上述《安全生产法》及《安全生产许可证条例》的相关规范外,还需遵守《药用辅料生产质量管理规范》《化学药物原料药制备和结构确证研究的技术指导原则》以及《化学药品技术标准》等具体行业要求。此外,如果医药中间体不直接涉及制药的核心环节,则在合规层面可能可以直接插入已有的GMP流程中,只要符合相应的要求和规范即可。就氨基酸及其衍生物产品而言,其与医药中间体类似,在不涉及特殊监管要求的情形下,符合安全生产的相关规定即可。

蔡 航|合伙人
业务领域:风险投资与私募股权、收购与兼并、医疗与生命科学

京若阳|律师
业务领域:私募股权投融资、并购重组、基金设立募集
邮箱:jingruoyang@anjielaw.com

王 菲 | 律师
业务领域:私募股权投融资、公司并购重组、合规
邮箱:wangfei@anjielaw.com

郁丹琳|律师助理
业务领域:私募股权投融资、公司并购重组、合规

王 婧| 律师助理
业务领域:私募股权投融资、合规
邮箱:wangjing@anjielaw.com
[1] 早在2005年,合成生物学产业化的先行者Amyris就研发了能够产生青蒿酸(artemisinic acid,合成青蒿素的重要前体)的酵母菌株。
[2] 相关数据为在Web of Science数据库、Medline数据库以及PubMed数据库中以“synthetic biology”和“drug”为检索词进行检索获得。
[3] 万秀坤等:《合成生物学发展现状与军事应用展望》,载《军事医学》2019年第11期。
[4] 值得一提的是,经美国专利商标局专利审判和上诉委员会(USPTO Patent Trial and Appeal Board)在今年2月裁决,在真核细胞中使用CRISPR基因编辑技术的专利归Broad Institute的张峰博士团队所有,而不是诺奖得主Jennifer Doudna 和 Emmanuelle Charpentier 所属的CVC团队。
[5] 专访丨“颠覆”医药研发,拥有“上帝视角”的合成生物学何以落地?https://new.qq.com/rain/a/20211111A06L4O00
[6] 2022年7月22日,国家卫生健康委就深化医改工作进展成效举行发布会,国家药监局药品监管司司长袁林在会上介绍了近几年国家药监局的工作内容,包括完善药品审评审批体系,制定发布相关制度规定和技术指南,推动和促进中药传承创新发展,深入推进医疗器械审评审批制度改革以及优化化妆品审评审批相关工作等内容。
[7] 例如金斯瑞(1548.HK)同时为合成生物学平台型企业及CGT CDMO(Contract Development and Manufacturing Organization,合同研发生产组织)企业。
[8] 截至2021年7月,我国批准上市的CGT药物为:上海三维的重组人5型腺病毒注射液(安柯瑞)、深圳赛百诺的重组人p53基因腺病毒颗粒(今又生)、复星凯特的CD19 CAR-T产品(基利仑赛注射液)和药明巨诺的CD19 CAR-T产品(瑞基奥仑赛注射液)。
[9] FDA《活体生物治疗产品的早期临床试验:化学、制造、和控制信息》产业指南,https://www.fda.gov/files/vaccines,%20blood%20&%20biologics/published/Early-Clinical-Trials-With-Live-Biotherapeutic-Products--Chemistry--Manufacturing--and-Control-Information--Guidance-for-Industry.pdf
[10] 参见《药品生产监督管理办法》政策解读,链接:https://www.nmpa.gov.cn/directory/web/nmpa/xxgk/zhcjd/zhcjdyp/20200331141001105.html。
[11] 根据《医疗器械监督管理条例》、《医疗器械生产监督管理办法》及《医疗器械经营监督管理办法》整理。
[12] 具体可参见《免于经营备案的第二类医疗器械产品目录》。
[13] 根据国家药品监督管理局1999年发布的《关于<药品生产企业许可证>换证工作的通知》中规定:对只生产药用化工原料、药用中间体、药用辅料、医用氧气的企业不列入换证范围。今后对上述产品的生产企业不再实施《药品生产企业许可证》管理。




























